
ALS and SMA are Motor Neuron Disorders
Spinal Muscular Atrophy (SMA) and Amyotrophic Lateral Sclerosis (ALS) are both Motor Neuron (MN) diseases, a group of progressive neurological disorders that destroy motor neurons. MNs are a special class of neurons that primarily connect the brain and spinal cord to the muscles, controlling voluntary movement and activities such as speaking, walking, breathing and swallowing. They are the longest cells of our body, reaching over a meter in length as they run through the spinal cord to the limb muscles.
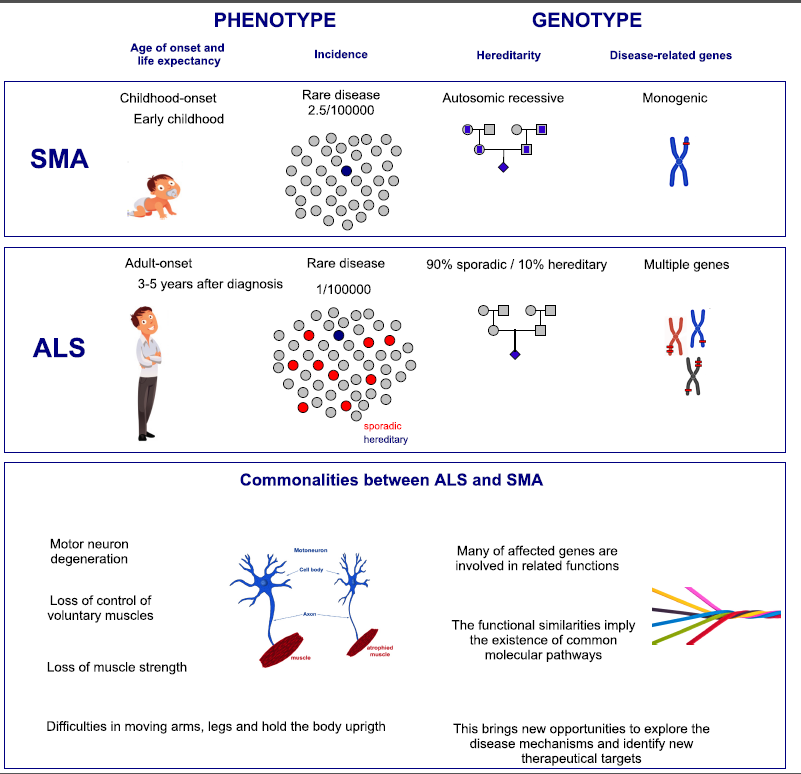
In general, individuals affected with ALS or SMA loose strength and the ability to move their arms and legs and hold the body upright. When the muscles of the diaphragm and chest wall fail to function properly, the ability to breathe without mechanical support is also lost, quickly leading to patient death.
SMA usually presents as a congenital hereditary disease in which, in the most severe cases, children never sit or stand, and die of respiratory failure before the age of two. On the other hand, ALS is usually diagnosed in adulthood (typically on the third decade of life), with most patients dying within 3 to 5 years from the onset of symptoms.
Although SMA is considered a rare disease, affecting to 1/100000 individuals, it is one of the most common genetic causes of death in children. On the other hand, ALS is the most frequent MN disorder, having a relatively high population incidence of 2.5/100000.
The molecular mechanisms causing MN degeneration remain unclear and there is still no effective treatment for these diseases. Research activities that contribute to the understanding of these mechanisms will provide innovative insights that can be translated into better diagnostics and therapeutic options for patients, eventually supporting the discovery of effective treatments or even a cure.
Why study ALS and SMA together?
Despite the known phenotypic and genotypic differences, there is an abundance of evidence supporting the existence of common molecular pathways operating in both disorders.
SMA is caused by low levels of the SMN protein, while ALS causing mutations occur in genes encoding the TDP-43 and FUS, among others. TDP-43, FUS and SMN interact with RNA molecules either directly or in the context of ribonucleoprotein particles (RNPs) and have been shown to regulate multiple steps in the gene expression pathway. Their functions include the regulation of transcription, splicing and microRNA biogenesis in the nucleus; and small nuclear RNP (snRNP) biogenesis, regulation of mRNA stability, translation and transport, predominantly in the cytoplasm. Although the connections between mutations in the genes encoding these proteins and neurodegeneration remain unresolved, alterations in neuronal RNA homeostasis (or ribostasis) are likely to represent a critical pathway in these diseases.
Consistent with a direct connection between ALS and SMA, TDP-43, FUS and SMN have been shown to physically and functionally interact. Overexpression of SMN, for example, rescues the axonal growth and branching defects observed in FUS mutant neurons. Furthermore, individuals with extra copies of the SMN gene have been reported to have an increased risk of developing ALS. These intricate connections suggest that ALS and SMA share common pathways that lead to MN degeneration.
From a Systems Medicine perspective, a disease can be viewed as an altered equilibrium state that involves the propagation of changes induced by a specific mutation through a complex network of interactions, moving the system away from its healthy state. Thus, different disease associated genes communicating at distant points of the network can lead to the same overall result. In the case of ALS and SMA, this is MN death. As a corollary, acting on critical network points, rather than directly targeting the disease causing genes, may restore the balance of the system back to a healthy state. By studying ALS and SMA together, the critical network hubs underlying MN degeneration may become easier to identify and thus lead to innovative therapeutic approaches.